Shape and functionality of nanoparticles
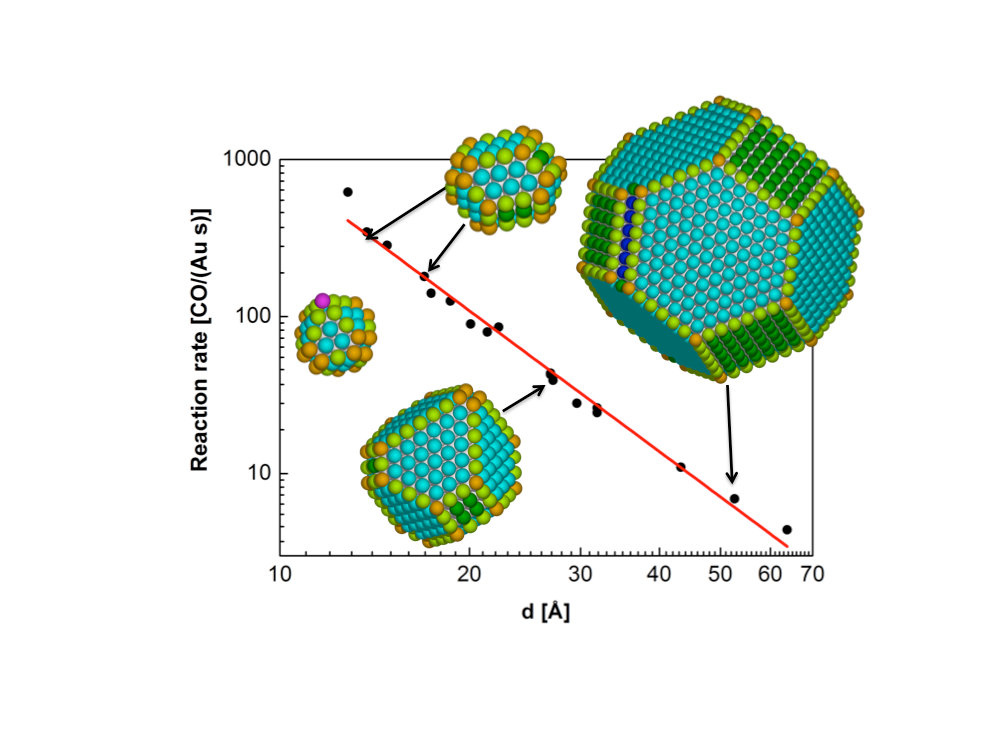
For many catalytics, the detailed atomic-scale shape of the nanoparticles matter, as the reaction almost exclusively occurs on special atomic sites, such as at the foot of a surface step or on corner atoms. In other reactions, the atomic-scale shape is less critical, instead the surface stress state may be important for the reaction rate. These effects are often dramatic, and may cause order of magnitude changes in the catalytic reaction rates.
We are developing multi-scale simulation methods to calculate the detailed state of nanoparticles including effects of support and environment. We couple these simulations with calculations of reaction rates of the active sites based on Density Functional Theory to predict the reactivity of potential catalytic materials, to improve the materials, and to design new catalytic materials.